mTOR: señalización y efectos en el cuerpo
La vía de señalización de la rapamicina (mTOR) de los mamíferos integra señales tanto intracelulares como extracelulares y sirve como un regulador central del metabolismo celular, el crecimiento, la proliferación y la supervivencia. Los descubrimientos realizados durante la última década muestran que la vía de mTOR se activa durante varios procesos celulares (p. Ej., Formación de tumores y angiogénesis, resistencia a la insulina, adipogénesis y activación de linfocitos T) y está desregulada en enfermedades humanas como el cáncer y la diabetes tipo 2. Estas observaciones han atraído un amplio interés científico y clínico en mTOR. Esto se destaca por el uso creciente de los inhibidores de mTOR [rapamicina y sus análogos (rapalogues)] en entornos patológicos, incluido el tratamiento de tumores sólidos, trasplante de órganos, reestenosis coronaria y artritis reumatoide.
¿Qué es mTOR?
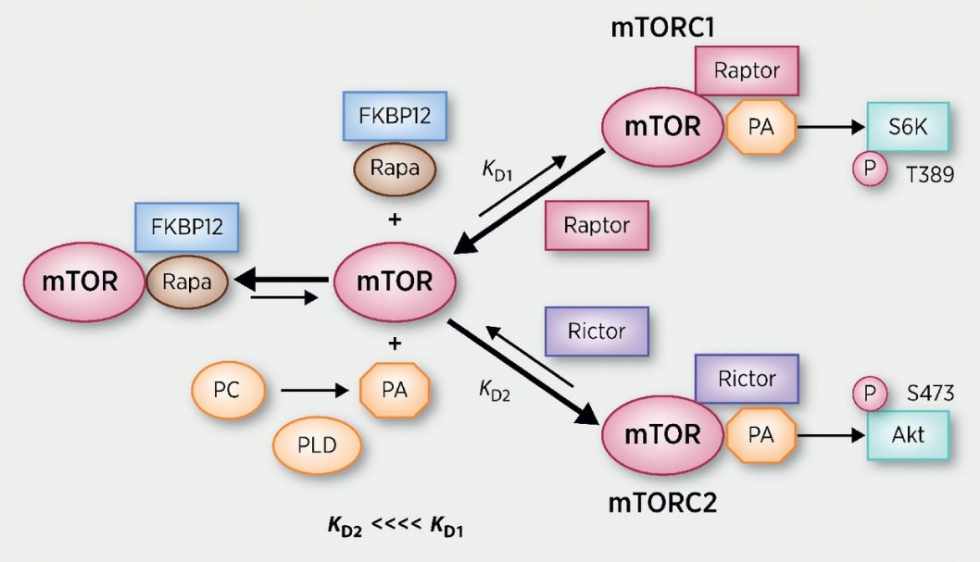
mTOR forma dos complejos funcionales, C1 y C2. El complejo C1 es más significativo en salud y enfermedad. mTOR responde a las señales de los nutrientes, los factores de crecimiento y el estado de la energía celular y controla el crecimiento y la proliferación celular en función de esto (mediante la regulación de la síntesis de proteínas).
En un lenguaje más simple, cuando tenemos mucha nutrición (principalmente proteínas) y calorías, esencialmente le decimos al cuerpo que los tiempos abundantes están aquí. Nuestras células aumentan su capacidad de trabajo y aumenta la producción de ATP. Las células aumentan la división y estamos preparados para el crecimiento y la reparación. mTOR es la proteína que detecta esto y pone «el pedal al metal».
Sin embargo, en momentos de estrés, como la reducción de la ingesta calórica o de nutrientes, se inhibe la mTOR (R).
Es importante darse cuenta de que mTOR puede aumentar el rendimiento y no debemos verlo como bueno o malo. Más bien depende.
Es decir, mTOR aumenta la producción de energía, pero también crea más productos basura.
La autofagia es el proceso que degrada estos productos basura. Pero solo es usualmente activo cuando mTOR disminuye. En otras palabras, el cuerpo no comienza a limpiar hasta que termina la fiesta.
La autofagia es equivalente a una «desintoxicación» desde una perspectiva científica. Necesitamos un equilibrio entre el crecimiento / productos basura y el descanso / limpieza.
Estructura mTOR y organización en complejos multiproteicos.
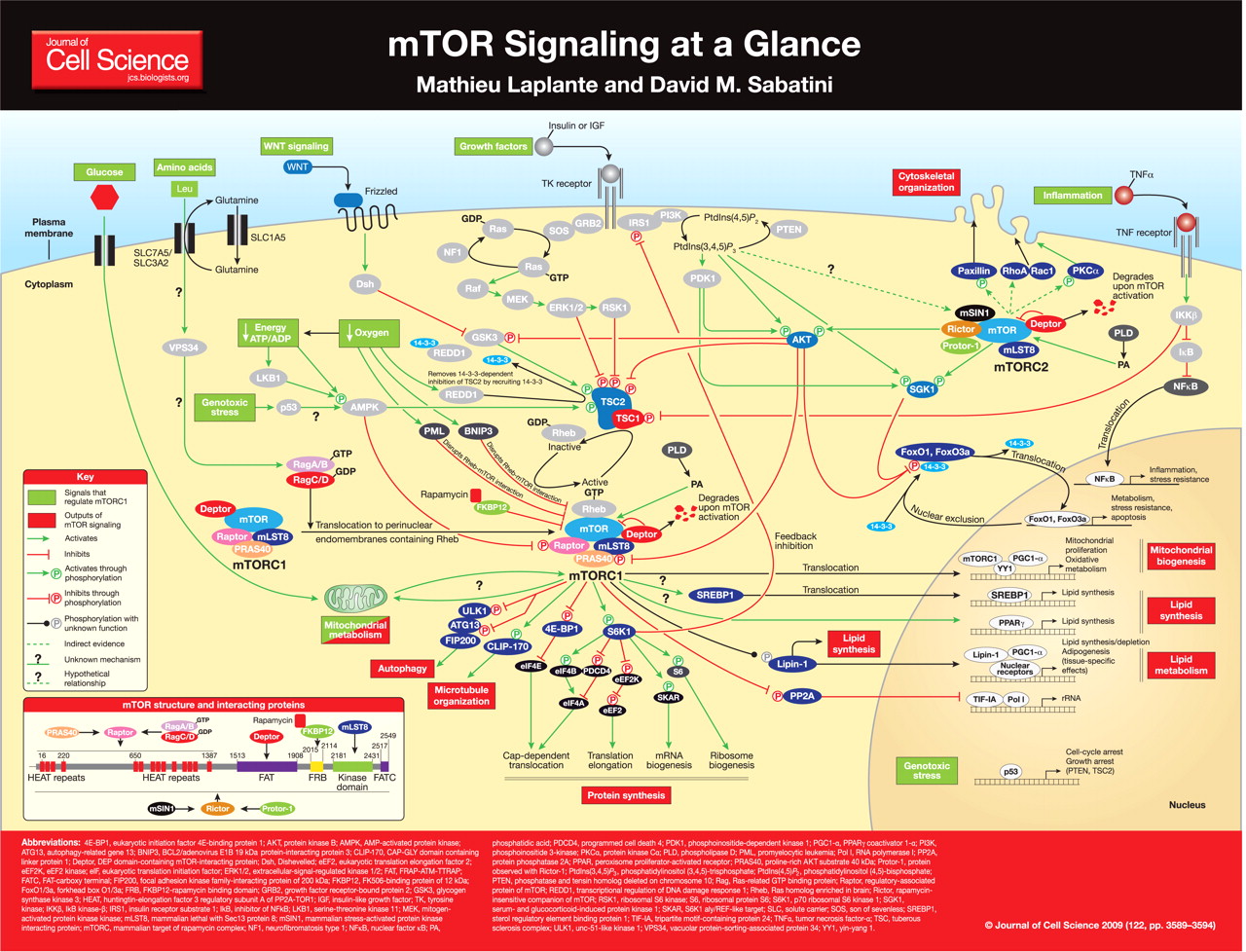
La proteína mTOR es una serina-treonina quinasa de 289 kDa que pertenece a la familia de quinasas relacionadas con fosfoinositido 3 quinasa (PI3K) y se conserva a lo largo de la evolución. El póster muestra una visión general de los dominios estructurales mTOR. mTOR nuclea al menos dos complejos multi-proteínas distintos, mTOR complex 1 (mTORC1) y mTOR complex 2 (mTORC2) (revisado por Guertin y Sabatini, 2007).
mTORC1
mTORC1 tiene cinco componentes: mTOR, que es la subunidad catalítica del complejo; proteína asociada al regulador de mTOR (raptor); mamífero letal con Sec13 proteína 8 (mLST8, también conocido como GβL); sustrato de AKT rico en prolina 40 kDa (PRAS40); y proteína que interactúa con mTOR que contiene el dominio DEP (Deptor) (Peterson et al., 2009). La función exacta de la mayoría de las proteínas que interactúan con mTOR en mTORC1 sigue siendo difícil de alcanzar. Se ha propuesto que Raptor podría afectar la actividad de mTORC1 al regular el ensamblaje del complejo y al reclutar sustratos para mTOR (Hara et al., 2002 ; Kim et al., 2002). El papel de mLST8 en la función mTORC1 tampoco está claro, ya que la eliminación de esta proteína no afecta la actividad de mTORC1 in vivo (Guertin et al., 2006). PRAS40 y Deptor se han caracterizado como distintos reguladores negativos de mTORC1 (Peterson et al., 2009 ; Sancak et al., 2007 ; Vander Haar et al., 2007). Cuando se reduce la actividad de mTORC1, PRAS40 y Deptor se reclutan en el complejo, donde promueven la inhibición de mTORC1. Se propuso que PRAS40 regula la actividad de la quinasa mTORC1 al funcionar como un inhibidor directo de la unión del sustrato (Wang et al., 2007). Tras la activación, mTORC1 fosforila directamente PRAS40 y Deptor, lo que reduce su interacción física con mTORC1 y activa aún más la señalización de mTORC1 (Peterson et al., 2009 ; Wang et al., 2007).
mTORC2
mTORC2 comprende seis proteínas diferentes, varias de las cuales son comunes a mTORC1 y mTORC2: mTOR; compañero de mTOR (rictor) insensible a la rapamicina; proteína que interactúa con la proteína quinasa activada por estrés de mamíferos (mSIN1); proteína observada con Rictor-1 (Protor-1); mLST8; y deptor. Existe alguna evidencia de que Rictor y mSIN1 se estabilizan entre sí, estableciendo la base estructural de mTORC2 (Frias et al., 2006 ; Jacinto et al., 2006). Rictor también interactúa con Protor-1, pero la función fisiológica de esta interacción no está clara (Thedieck et al., 2007 ; Woo et al., 2007). De manera similar a su papel en mTORC1, Deptor regula negativamente la actividad de mTORC2 (Peterson et al., 2009); Hasta el momento, Deptor es el único inhibidor endógeno caracterizado de mTORC2. Finalmente, mLST8 es esencial para la función mTORC2, ya que la eliminación de esta proteína reduce severamente la estabilidad y la actividad de este complejo (Guertin et al., 2006).
Ahora que se han identificado muchas proteínas que interactúan con mTOR, se necesitarán estudios bioquímicos adicionales para aclarar las funciones de estas proteínas en la señalización de mTOR y sus posibles implicaciones en la salud y la enfermedad. A continuación, discutimos la comprensión actual de las funciones de mTORC1 y mTORC2.
mTORC1: un regulador maestro del crecimiento celular y el metabolismo
mTORC1 regula positivamente el crecimiento y la proliferación celular al promover muchos procesos anabólicos, incluida la biosíntesis de proteínas, lípidos y orgánulos, y al limitar los procesos catabólicos como la autofagia. Gran parte del conocimiento sobre la función mTORC1 proviene del uso de la macramida rapamicina bacteriana. Al ingresar a la célula, la rapamicina se une a la proteína de unión a FK506 de 12 kDa (FKBP12) e interactúa con el dominio de unión a FKBP12-rapamicina (FRB) de mTOR, inhibiendo así las funciones de mTORC1 (revisado por Guertin y Sabatini, 2007). En contraste con su efecto sobre mTORC1, la FKBP12-rapamicina no puede interactuar físicamente con o inhibir de manera aguda mTORC2 (Jacinto et al., 2004 ; Sarbassov et al., 2004). Sobre la base de estas observaciones, mTORC1 y mTORC2 se han caracterizado respectivamente como complejos sensibles a la rapamicina e insensibles a la rapamicina. Sin embargo, este paradigma podría no ser totalmente exacto, ya que el tratamiento crónico con rapamicina puede, en algunos casos, inhibir la actividad de mTORC2 al bloquear su ensamblaje (Sarbassov et al., 2006). Además, informes recientes sugieren que las funciones importantes de mTORC1 son resistentes a la inhibición de la rapamicina (Choo et al., 2008 ; Feldman et al., 2009 ; Garcia-Martinez et al., 2009 ; Thoreen et al., 2009).
Síntesis de proteínas
mTORC1 controla positivamente la síntesis de proteínas, que se requiere para el crecimiento celular, a través de varios efectores posteriores. mTORC1 promueve la síntesis de proteínas mediante la fosforilación de la proteína 1 de unión al factor de iniciación eucariótico 4E (eIF4E) (4E-BP1) y la quinasa 1 ribosomal p70 (S6K1) p70. La fosforilación de 4E-BP1 evita su unión a eIF4E, lo que permite que eIF4E promueva la traducción dependiente del límite (revisado por Richter y Sonenberg, 2005). La estimulación de la actividad de S6K1 por mTORC1 conduce a aumentos en la biogénesis del ARNm, la traducción y el alargamiento dependientes de los capuchones, y la traducción de proteínas ribosómicas a través de la regulación de la actividad de muchas proteínas, como el objetivo S6K1 aly / REF (SKAR), programada muerte celular 4 (PDCD4), factor de alargamiento eucariota 2 quinasa (eEF2K) y proteína ribosomal S6 (revisada porMa y Blenis, 2009). También se ha demostrado que la activación de mTORC1 promueve la biogénesis de los ribosomas estimulando la transcripción del ARN ribosómico a través de un proceso que involucra la proteína fosfatasa 2A (PP2A) y el factor de iniciación de la transcripción IA (TIF-IA) (Mayer et al., 2004).
Autofagia
La autofagia, es decir, el secuestro de componentes intracelulares dentro de los autofagosomas y su degradación por los lisosomas, es un proceso catabólico que es importante en la degradación de los orgánulos y el recambio de proteínas. Cuando la disponibilidad de nutrientes es limitada, la degradación de orgánulos y complejos de proteínas a través de la autofagia proporciona material biológico para sostener procesos anabólicos como la síntesis de proteínas y la producción de energía. Los estudios han demostrado que la inhibición de mTORC1 aumenta la autofagia, mientras que la estimulación de mTORC1 reduce este proceso (revisado por Codogno y Meier, 2005). Hemos observado que mTORC1 controla la autofagia a través de un mecanismo desconocido que es esencialmente insensible a la inhibición por la rapamicina (Thoreen et al., 2009). Recientemente, tres grupos independientes demostraron que mTORC1 controla la autofagia mediante la regulación de un complejo de proteínas compuesto por unc-51-like kinasa 1 (ULK1), el gen 13 relacionado con la autofagia (ATG13) y la proteína de interacción de la familia de la quinasa de adhesión focal de 200 kDa (FIP200) (Ganley et al., 2009 ; Hosokawa et al., 2009 ; Jung et al., 2009). Estos estudios han revelado que mTORC1 reprime la autofagia mediante la fosforilación y, por lo tanto, reprime a ULK1 y ATG13.
Síntesis lipídica
El papel de mTORC1 en la regulación de la síntesis de lípidos, que se requiere para el crecimiento y la proliferación celular, está comenzando a apreciarse. Se ha demostrado que mTORC1 regula positivamente la actividad de la proteína de unión al elemento regulador del esterol 1 (SREBP1) (Porstmann et al., 2008) y del receptor γ activado por el proliferador de peroxisoma (PPARγ) (Kim y Chen, 2004), dos transcripciones. Factores que controlan la expresión de genes que codifican proteínas involucradas en la homeostasis de lípidos y colesterol. El bloqueo de mTOR con rapamicina reduce la expresión y la actividad de transactivación de PPARγ (Kim y Chen, 2004). El mecanismo molecular de la activación de SREBP1 por mTORC1 es desconocido. Además, la rapamicina reduce la fosforilación de la lipina-1 (Huffman et al., 2002), una fosfatasa de ácido fosfatídico (PA) que participa en la síntesis de glicerolípidos y en la coactivación de muchos factores de transcripción relacionados con el metabolismo de los lípidos, incluidos PPARγ, PPARα y PGC1-α. El impacto preciso de la fosforilación de la lipina-1 en la síntesis de lípidos aún no se ha establecido.
Metabolismo mitocondrial y biogénesis.
El metabolismo mitocondrial y la biogénesis están regulados por mTORC1. La inhibición de mTORC1 por la rapamicina reduce el potencial de membrana mitocondrial, el consumo de oxígeno y los niveles celulares de ATP, y altera profundamente el fosfoproteoma mitocondrial (Schieke et al., 2006). Recientemente, se ha observado que el número de copias del ADN mitocondrial, así como la expresión de muchos genes que codifican proteínas involucradas en el metabolismo oxidativo, se reducen con la rapamicina y aumentan con las mutaciones que activan la señalización de mTORC1 (Chen et al., 2008 ; Cunningham et al., 2007). Además, la eliminación condicional de Raptor en el músculo esquelético del ratón reduce la expresión de los genes implicados en la biogénesis mitocondrial (Bentzinger et al., 2008). Cunningham y sus colegas han descubierto que mTORC1 controla la actividad transcripcional del coactivador 1 (PGC1-α) de PPARγ, un cofactor nuclear que desempeña un papel clave en la biogénesis mitocondrial y el metabolismo oxidativo, al alterar directamente su interacción física con otro factor de transcripción, a saber, yin yang 1 (YY1) (Cunningham et al., 2007).
Muchos caminos conducen a mTORC1: descripción general de una red de señalización compleja
mTORC1 integra cuatro señales principales (factores de crecimiento, estado energético, oxígeno y aminoácidos) para regular muchos procesos que participan en la promoción del crecimiento celular. Uno de los sensores más importantes involucrados en la regulación de la actividad de mTORC1 es el complejo de esclerosis tuberosa (TSC), que es un heterodímero que comprende TSC1 (también conocido como hamartina) y TSC2 (también conocido como tuberina). TSC1 / 2 funciona como una proteína activadora de GTPasa (GAP) para la pequeña GTPasa Rheb relacionada con Ras (homólogo de Ras enriquecido en el cerebro). La forma activa, unida a GTP de Rheb interactúa directamente con mTORC1 para estimular su actividad (Long et al., 2005 ; Sancak et al., 2007). El mecanismo exacto por el cual Rheb activa mTORC1 queda por determinar. Como un GAP específico de Rheb, TSC1 / 2 regula negativamente la señalización de mTORC1 convirtiendo a Rheb en su estado inactivo unido al PIB (Inoki et al., 2003 ; Tee et al., 2003). Consistente con el papel de TSC1 / 2 en la regulación negativa de mTORC1, las mutaciones inactivantes o la pérdida de heterocigosidad de TSC1 / 2 dan lugar a esclerosis tuberosa, una enfermedad asociada con la presencia de numerosos tumores benignos que están compuestos por células agrandadas y desorganizadas (revisado por Crino et al., 2006).
Factores de crecimiento
Los factores de crecimiento estimulan mTORC1 a través de la activación de la insulina canónica y las vías de señalización de Ras. La estimulación de estas vías aumenta la fosforilación del TSC2 por la proteína quinasa B (PKB, también conocida como AKT) (Inoki et al., 2002 ; Potter et al., 2002), por la quinasa 1/2 regulada por señales extracelulares (ERK1 / 2) (Ma et al., 2005), y p90 ribosomal S6 quinasa 1 (RSK1) (Roux et al., 2004), y conduce a la inactivación de TSC1 / 2 y, por tanto, a la activación de mTORC1. Además, la activación de AKT por factores de crecimiento puede activar mTORC1 de manera independiente de TSC1 / 2 al promover la fosforilación y disociación de PRAS40 de mTORC1 (Sancak et al., 2007 ; Vander Haar et al., 2007; Wang et al., 2007).
La unión de la insulina a su receptor de la superficie celular promueve la actividad tirosina quinasa del receptor de insulina, el reclutamiento del sustrato 1 del receptor de insulina (IRS1), la producción de fosfatidilinositol (3,4,5) -trifosfato [PtdIns (3,4, 5) P 3] a través de la activación de PI3K, y el reclutamiento y activación de AKT en la membrana plasmática. En muchos tipos de células, la activación de mTORC1 reprime fuertemente el eje PI3K-AKT en sentido ascendente de PI3K. La activación de S6K1 por mTORC1 promueve la fosforilación de IRS1 y reduce su estabilidad (revisado por Harrington et al., 2005). Se ha demostrado que esta vía de autorregulación, caracterizada por el circuito de retroalimentación negativa dependiente de S6K1, tiene profundas implicaciones tanto para las enfermedades metabólicas como para la tumorigénesis (revisada porManning, 2004). También es probable que otras vías que son independientes de IRS1 contribuyan a la retroinhibición de mTORC1. Por ejemplo, la pérdida de TSC1 / 2 suprime la expresión del receptor del factor de crecimiento derivado de plaquetas (PDGFR) de una manera sensible a la rapamicina (Zhang et al., 2007). Cómo la señalización de mTOR controla la expresión de PDGFR queda por determinar.
Estado energético
El estado energético de la célula se señala a mTORC1 a través de la proteína quinasa activada por AMP (AMPK), un sensor maestro del estado energético intracelular (revisado por Hardie, 2007). En respuesta al agotamiento de la energía (baja relación ATP: ADP), AMPK se activa y fosforila el TSC2, lo que aumenta la actividad GAP del TSC2 hacia Rheb y reduce la activación de mTORC1 (Inoki et al., 2003). Además, AMPK puede reducir la actividad de mTORC1 en respuesta al agotamiento de la energía mediante la fosforilación directa de Raptor (Gwinn et al., 2008).
Niveles de oxigeno
Los niveles de oxígeno afectan la actividad de mTORC1 a través de múltiples vías (revisado por Wouters y Koritzinsky, 2008). Bajo condiciones de hipoxia leve, la reducción en los niveles de ATP activa AMPK, que promueve la activación de TSC1 / 2 e inhibe la señalización de mTORC1 como se describe en la sección anterior (Arsham et al., 2003 ; Liu et al., 2006). La hipoxia también puede activar TSC1 / 2 a través de la regulación transcripcional de la respuesta de daño al ADN 1 (REDD1) (Brugarolas et al., 2004 ; Reiling y Hafen, 2004). REDD1 bloquea la señalización de mTORC1 mediante la liberación de TSC2 de su asociación inducida por el factor de crecimiento con proteínas 14-3-3 (DeYoung et al., 2008). Esta capacidad de REDD1 para reducir la señalización de mTORC1 al interrumpir la interacción de TSC2 y 14-3-3 probablemente ha evolucionado para limitar los procesos que consumen energía cuando el oxígeno, pero no los factores de crecimiento, es escaso. Además, el supresor de tumores de leucemia promielocítica (PML) y la proteína 3 que interactúa con la proteína BCL2 / adenovirus E1B 19 kDa (BNIP3) reducen la señalización de mTORC1 durante la hipoxia al interrumpir la interacción entre mTOR y su regulador positivo Rheb (Bernardi et al., 2006 ; Li et al. al., 2007).
Aminoácidos
Los aminoácidos representan una señal fuerte que regula positivamente mTORC1 (revisado por Guertin y Sabatini, 2007). Recientemente se demostró que la leucina, un aminoácido esencial requerido para la activación de mTORC1, se transporta a las células de una manera dependiente de la glutamina (Nicklin et al., 2009).). La glutamina, que se importa a las células a través de SLC1A5 [miembro de la familia de portadores de soluto 1 (transportador de aminoácidos neutros) 5], se intercambia para importar leucina a través de un sistema heterodimérico compuesto por SLC7A5 [familia de portadores de solutos de antipuerto 7 (transportador de aminoácidos catiónicos, sistema y +, miembro 5] y SLC3A2 [familia portadora de solutos 3 (activadores del transporte de aminoácidos neutros y dibásicos) miembro 2]. El mecanismo por el cual los aminoácidos intracelulares luego se señalizan a mTORC1 permaneció oscuro durante muchos años. La activación de mTORC1 por aminoácidos es Se sabe que son independientes de TSC1 / 2, debido a que la ruta mTORC1 sigue siendo sensible a la privación de aminoácidos en células que carecen de TSC1 o TSC2 (Nobukuni et al., 2005). Algunos estudios han implicado la proteína asociada a la clasificación de la proteína vacuolar humana 34 (VPS34)) en la detección de nutrientes (Nobukuni et al., 2005); sin embargo, el rol preciso del VPS34 humano en este proceso aún no se ha establecido (Juhasz et al., 2008).
Recientemente, dos equipos independientes, incluido el nuestro, han demostrado que las proteínas Rag, una familia de cuatro pequeñas GTPasas relacionadas, interactúan con mTORC1 de una manera sensible a los aminoácidos y son necesarias para la activación de la ruta de los mTORC1 por los aminoácidos (Kim et al. al., 2008 ; Sancak et al., 2008). En presencia de aminoácidos, las proteínas Rag se unen a Raptor y promueven la relocalización de mTORC1 desde ubicaciones discretas en todo el citoplasma a una región perinuclear que contiene su activador Rheb (Sancak et al., 2008). La disociación física de mTORC1 y Rheb con privación de aminoácidos podría explicar por qué los activadores de Rheb, como los factores de crecimiento, no pueden estimular la señalización de mTORC1 en ausencia de aminoácidos.
Otras condiciones y señales celulares.
Además de las señales clave descritas anteriormente, se ha demostrado que otras condiciones y señales celulares, como el estrés genotóxico, la inflamación, el ligando Wnt y la PA, regulan la señalización de mTORC1. El estrés genotóxico reduce la actividad mTORC1 a través de muchos mecanismos. Por ejemplo, la activación de p53 en respuesta al daño en el ADN activa rápidamente la AMPK a través de un proceso desconocido, que a su vez fosforila y, por lo tanto, activa el TSC2 (Feng et al., 2005). Además, p53 controla negativamente la señalización de mTORC1 al aumentar la transcripción de la fosfatasa y el homólogo de tensina eliminado en el cromosoma 10 (PTEN) y TSC2, dos reguladores negativos de la vía (Feng et al., 2005 ; Stambolic et al., 2001). Los mediadores inflamatorios también envían señales a mTORC1 a través del complejo TSC1 / 2. Las citoquinas proinflamatorias, como el TNFα, activan la IκB cinasa-β (IKKβ), que interactúa físicamente con TSC1 y lo desactiva, lo que lleva a la activación de mTORC1 (Lee et al., 2007). Se cree que esta relación positiva entre la inflamación y la activación de mTORC1 es importante en la angiogénesis tumoral (Lee et al., 2007) y en el desarrollo de resistencia a la insulina (Lee et al., 2008). La señalización Wnt también aumenta la actividad de mTORC1 a través de la inactivación de TSC1 / 2. La estimulación de la vía Wnt inhibe la glucógeno sintasa quinasa 3 (GSK3), una quinasa que promueve la actividad de TSC1 / 2 mediante la fosforilación directa de TSC2 (Inoki et al., 2006).). Finalmente, PA ha sido identificado como otro activador de mTORC1. Muchos grupos han demostrado que la PA exógena o la sobreexpresión de enzimas productoras de PA como la fosfolipasa D1 (PLD1) y PLD2 aumentan significativamente la señalización de mTORC1 (revisado por Foster, 2007). Un estudio reciente sugiere que la PA afecta la señalización de mTOR facilitando el ensamblaje de complejos de mTOR, o estabilizando los complejos (Toschi et al., 2009).
mTORC2 todavía tiene muchos secretos por revelar
En contraste con mTORC1, para el cual se han definido muchas señales en sentido ascendente y funciones celulares (ver arriba), se sabe relativamente poco acerca de la biología de mTORC2. La letalidad temprana causada por la eliminación de los componentes de mTORC2 en ratones, así como la ausencia de inhibidores de mTORC2, ha complicado el estudio de este complejo de proteínas. No obstante, se han hecho muchos descubrimientos importantes en los últimos años. Utilizando diversos enfoques genéticos, se ha demostrado que mTORC2 desempeña funciones clave en diversos procesos biológicos, incluida la supervivencia celular, el metabolismo, la proliferación y la organización del citoesqueleto. El papel de mTORC2 en estos procesos se discute con más detalle a continuación.
Supervivencia celular, metabolismo y proliferación.
La supervivencia celular, el metabolismo y la proliferación dependen en gran medida del estado de activación de AKT, que regula positivamente estos procesos a través de la fosforilación de varios efectores (revisado por Manning y Cantley, 2007). La activación completa de AKT requiere su fosforilación en dos sitios: Ser308, por la quinasa 1 dependiente de fosfoinositida (PDK1), y Ser473, por una quinasa que no se identificó durante muchos años, pero nuestro grupo demostró ser mTORC2 en 2005 (Sarbassov et al., 2005). Otros estudios han observado posteriormente que la ablación de varios componentes mTORC2 bloquea específicamente la fosforilación de AKT en Ser473 y la fosforilación en sentido descendente de algunos, pero no todos, sustratos de AKT (Guertin et al., 2006 ; Jacinto et al., 2006). La inhibición de la AKT después del agotamiento de mTORC2 reduce la fosforilación y, por lo tanto, activa, los factores de transcripción de la caja de cabeza de tenedor O1 (FoxO1) y FoxO3a, que controlan la expresión de genes implicados en la resistencia al estrés, el metabolismo, la detención del ciclo celular y la apoptosis (revisada por Calnan y Brunet, 2008). Por el contrario, el estado de fosforilación de TSC2 y GSK3 no se ve afectado por la inactivación de mTORC2. Recientemente, la proteína quinasa 1 inducida por suero y glucocorticoides (SGK1), que comparte homología con AKT, también demostró estar regulada por mTORC2 (García-Martínez y Alessi, 2008). En contraste con el AKT, que conserva una actividad basal cuando se inhibe el mTORC2, la actividad de SGK1 se anula totalmente en estas condiciones. Debido a que SGK1 y AKT fosforilan a FoxO1 y FoxO3a en sitios comunes, es posible que la falta de actividad de SGK1 en células deficientes en mTORC2 sea responsable de la inhibición de la fosforilación de FoxO1 y FoxO3a.
Organización citoesquelética
mTORC2 regula la organización del citoesqueleto. Muchos grupos independientes han observado que derribar los componentes de mTORC2 afecta la polimerización de la actina y perturba la morfología celular (Jacinto et al., 2004 ; Sarbassov et al., 2004). Estos estudios han sugerido que mTORC2 controla el citoesqueleto de actina mediante la promoción de la fosforilación de la proteína quinasa Cα (PKCα), la fosforilación de la paxilina y su relocalización a adherencias focales, y la carga de GTP de RhoA y Rac1. El mecanismo molecular por el cual mTORC2 regula estos procesos no se ha determinado.
Señalización a mTORC2: la caja negra
Las vías de señalización que conducen a la activación de mTORC2 no están bien caracterizadas. Debido a que los factores de crecimiento aumentan la actividad de quinasa mTORC2 y la fosforilación de AKT en Ser473, se considera que son una señal plausible para regular esta vía (revisado por Guertin y Sabatini, 2007). Con la estimulación del factor de crecimiento, el AKT se fosforila en la membrana celular a través de la unión de PtdIns (3,4,5) P 3 a su dominio de homología de pleckstrin (PH). Bajo estas condiciones, PDK1 también se recluta a la membrana a través de su dominio PH y fosforila AKT en Ser308 (revisado por Lawlor y Alessi, 2001).). Curiosamente, el componente mTORC2 mSIN1 posee un dominio PH en su extremo C, lo que sugiere que mSIN1 puede promover la translocación de mTORC2 a la membrana y la fosforilación de AKT en Ser473. Se necesita trabajo adicional para respaldar este modelo e identificar otras señales celulares que desempeñan un papel en la regulación de mTORC2.
mTOR: efectos negativos
Demasiada activación de mTOR contribuye a un gran número de enfermedades humanas, como el cáncer, la obesidad, la diabetes tipo 2, la depresión y la neurodegeneración (R). También puede ser responsable del acné (R).
Para dar un ejemplo del mundo real, recientemente tuve un cliente que era sensible a la lectina y se sometió a una dieta tipo paleo de carne y verduras. Todavía estaba teniendo problemas con la inflamación. Su testosterona y sus hormonas estaban por las nubes. Después de armar una foto, le pregunté si se ponía el músculo con mucha facilidad. Dijo que lo hizo y que todos sus amigos estaban celosos de que apenas hizo ejercicio y tenía buena musculatura. Resulta que él también tiene problemas graves con el acné. Sobre la base de esta imagen, la sobreactivación de mTOR es una buena hipótesis. Tiene una ingesta de proteínas muy alta y experimenta inflamación, acné, depresión, crecimiento muscular fácil y hormonas elevadas.
El mTOR está asociado con el cáncer y, de hecho, aumenta la angiogénesis (a través de HIF-1a), un proceso a través del cual se forman nuevos vasos sanguíneos a partir de vasos preexistentes. (R) Esto ayuda al crecimiento del cáncer.
El aumento de mTOR promueve la inmunidad Th1 y Th17, lo que lleva a un aumento de la inflamación intestinal (R), entre otras cuestiones. Aumenta las células Th17 al aumentar otra proteína llamada hipoxia inducida por factor (HIF) -1α. (R)
Una reducción de mTOR Mejora la sensibilidad a la insulina en las células musculares. (R)
Técnico: mTOR aumenta la glucólisis, que es lo que permite que las células Th17 proliferen. Esto funciona a través de HIF1α. El bloqueo de la glucólisis inhibida Th 17 de desarrollo, mientras que la promoción de T reg células generación. (R)
Cuando las células T (CD4 y CD8) son estimuladas, por lectinas u otros medios, se reproducen rápidamente. (R)
La rápida producción de células T requiere energía. La activación de mTOR permite que las células T se expandan rápidamente al cambiar la forma en que obtienen energía. En lugar de obtener energía de las mitocondrias (a través de la fosforilación oxidativa), la obtienen principalmente al descomponer la glucosa (glucólisis). (R)
Cuando impides este proceso de descomposición de la glucosa, las células T se dan cuenta de que no tienen lo necesario para expandirse rápidamente y combatir los patógenos. Así que, en lugar de eso, se convierten en células Treg, que controlan el sistema inmunológico. (R)
Esta es una buena imagen que muestra las condiciones necesarias para estas cuatro células T.
mTOR: efectos positivos
La activación de mTOR nos permite poner más músculo (y grasa) (R) y aumentamos varias hormonas como IGF-1 (R). Si eres demasiado musculoso (R), sospecho que mTOR hiperactivo.
Esto tiene sentido evolutivo. Cuando teníamos comida, era una buena idea aumentar los músculos y la grasa y, cuando no lo teníamos, tenía sentido apagar nuestros sistemas para conservar energía.
En ratas, se ha demostrado que la activación de mTOR en el hipotálamo disminuye la ingesta de alimentos y el peso corporal. La leptina causa saciedad por este mecanismo (R). El NPY (aumenta el hambre) aumenta cuando se inhibe mTOR, lo que también sugiere que la activación de mTOR inhibirá el apetito (R). Confusamente, otro estudio más reciente dice que la grelina causa la activación de mTOR hipotalámico e inhibe el hambre inhibida por mTOR. (R) ¿Tal vez mTOR bajo o alto inhibe el hambre?
mTOR aumenta la producción de ATP y crea nuevas mitocondrias . (R) También aumenta el metabolismo mitocondrial (al activar PGC1a). (R)
mTOR está involucrado en varias formas de plasticidad sináptica y consolidación de la memoria. La inhibición de mTOR puede ser útil en personas con trastorno de estrés postraumático porque bloquea la reconsolidación de una memoria de miedo establecida de manera duradera (R).
Sin embargo, la sobreactivación de mTOR también causa defectos en la plasticidad y la memoria (R).
El punto clave
Para la salud y la longevidad, queremos que los niveles sistémicos de mTOR se encuentren por debajo de la mayoría del tiempo, con ataques de activación.
Es preferible tener mTOR más activo en su cerebro y músculos que en sus células de grasa e hígado. El ejercicio es ideal porque hace exactamente esto. (R)
Enfermedades asociadas con la activación de mTOR
Esta no es una lista completa.
- Envejecimiento (R): se supone que la restricción calórica y la restricción de metionina causan la extensión de la vida útil al disminuir la actividad de mTOR
- Cáncer (R) – Mama (R)
- Enfermedad autoinmune – aumenta Th1 y Th17
- Depresión (R)
- Diabetes (R),
- ¿Obesidad (R), causa o efecto?
- Alzheimer (R),
- Degeneración macular (R),
- Enfermedad del riñón (R),
- Epilepsia (R),
- Autismo (R): mTOR previene la «poda» o la «autofagia» de las sinapsis excitatorias en los trastornos del espectro autista. (R)
- Dolor crónico (R),
- LES (R)
Activadores de mTOR
El principal activador de mTOR es una variedad de aminoácidos y la hormona insulina. La testosterona también es capaz de activar mTOR (R, R2).
- Proteínas, especialmente leucina.
- Exceso de calorías
- Exceso de carbohidratos
- Ejercicio (R, R2): activado en el cerebro, los músculos y el corazón… Inhibido en el hígado y las células grasas. Todo bien…
- Orexin (R)
- IGF-1 (R)
- Insulina
- Testosterona (R)
- Ghrelin (R) – en el hipotálamo
- Leptina (R) – en el hipotálamo
- Hormona tiroidea (R): en el hipotálamo… y otras células (R)
- Oxígeno
- Ketamina (R). (En el cerebro – produce efecto antidepresivo.)
- IL-6 (R) – en músculo y grasa
Inhibidores naturales de mTOR
Dado que la restricción de proteínas, el resveratrol, la curcumina, el EGCG y la metformina inhiben la mTOR de diferentes maneras, apuesto a que tomarlas juntas creará una inhibición grave de la mTOR.
- Restricción de proteínas (R, R2): Restricción de leucina (R), ¿Restricción de glutamina? (R), restricción de metionina ? (R), restricción de lisina ? (R), restricción de arginina ? (R), restricción de treonina (R), restricción de isoleucina (R),
- ¿Glutamina? (R)
- Restricción calórica (R),
- Dietas Ketogénicas (R)
- Restricción calórica intermitente (R),
- Ejercicio (R, R2) – Inhibido en células de hígado y grasa. Activado en cerebro, músculo y corazón… todo bien…
- Cortisol / Glucocorticoides (R)
- Metformina (R), (al mejorar la asociación de PRAS40 con RAPTOR)
- NAC (R) (Ensayo clínico: «actividad mTOR profundamente reducida» en células T)
- Resveratrol (R) (C1 / C2), único) aumentó la asociación entre mTOR y su inhibidor, DEPTOR. (R)
- Aspirina (R):células de cáncer colorrectal,
- Hígado de bacalao / Omega-3 (R) (C1 / C2),
- Aceite de oliva virgen extra (R)
- EGCG / té (R, R2) (C1 /?) – Inhibidor competitivo de ATP de PI3K y mTOR (R)
- Curcumina (R, R2) (C1 / C2)…. mecanismo único – separa a la rapaz de mTOR (R)
- Ácido R-lipoico (R) – también disminuyó la quinasa p70S6 (R)
- Cafeína (R, R2, R3) (C1 /?),
- Fisetin (R) – células de grasa,
- Apigenina (R) (AMPK +, Akt -)
- Quercetina (R), (PI3K / Akt -, AMPK +, Hamartin +)
- Genistein (R),
- DIM (R),
- Ácido ursólico (R),
- Alcohol (R) (C1 / C2),
- Emodin (que se encuentra en Fo-Ti, Resveratrol, Ruibarbo, Aloe,) (R) (C2),
- Andrographis / Andrographolide (R), (PI3K / Akt-)
- Granada / ácido elágico (R),
- Reishi (R)
- Cardo mariano / silimarina (R),
- Ácido oleanólico (R) (C1),
- Antocianinas / Extracto de semilla de uva (R),
- Astrágalo (R), (cáncer de colon)
- Rhodiola (R)
- Carnosine (R)
- Plumbagin (casco de nogal negro) (R), Glucagon (R), AICAR (R)
- Vías: IGF (-)… PI3K (-)… Akt (-)… AMPK (+)…
Activadores AMPK inhiben mTOR
La activación de AMPK da como resultado la reducción de mTOR.
Pero puede tener escenarios en los que tanto AMPK está activado como mTOR también está activado porque AMPK no lo inhibe directamente; Inhibe otra proteína que aumenta directamente la mTOR. Por ejemplo, Ghrelin, la hormona del hambre, activa AMPK y mTOR en el hipotálamo. (R)
Superóxido Dismutasa 2 (SOD): función, niveles, suplementos

Colágeno ¿cuál es su funcionalidad en el cuerpo humano?

Plantibacter sp. Leaf314: Descubrimientos recientes
Sobre el autor
saludybelleza
Rafa Montes, editor de Salud y Belleza. Investigación de temas de salud, bienestar, psicología y más. Puedes encontrarme en LinkedIn