Mitocondria: función y enfermedades asociadas
Las mitocondrias son la potencia de la célula. Son la parte de la célula responsable de convertir los alimentos en energía para el cuerpo. La disfunción mitocondrial está vinculada a una serie de enfermedades. Lea esta publicación para saber todo lo que necesita saber sobre las mitocondrias, cómo funcionan y cómo pueden afectar su salud.
¿Qué es la mitocondria?
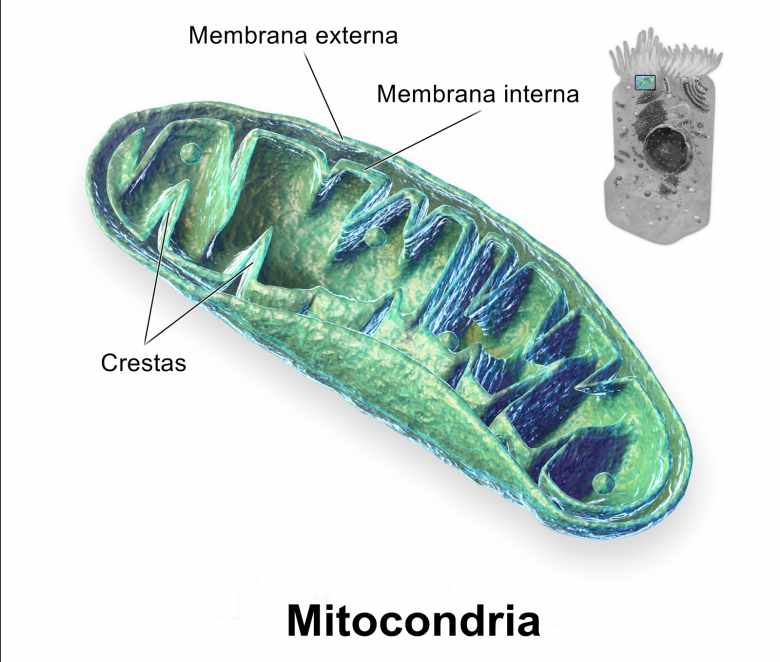
La mitocondria (mitocondria plural) es un orgánulo cerrado por membrana responsable de la conversión de grasas y carbohidratos (glucosa) en formas utilizables de energía para el cuerpo [R, R].
La función principal de las mitocondrias es producir ATP, la principal fuente de energía utilizada por todas las células. El ATP se utiliza para todas las actividades necesarias de la célula, y debemos comer para reponer la producción de ATP [R].
Las mitocondrias usan oxígeno para producir ATP en un proceso conocido como respiración aeróbica o celular. Esta reacción utiliza oxígeno y produce dióxido de carbono, que exhalamos a través de nuestros pulmones [R].
El número de mitocondrias por célula varía con cada tipo de célula. A medida que aumenten las demandas de energía de una célula, por ejemplo, en las células musculares, también aumentará el número de mitocondrias. Esto permite una mayor producción y uso de energía en las células activas [R].
Aparte de la producción de energía, las mitocondrias desempeñan un papel vital en el monitoreo de los niveles de calcio, lo que ayuda a equilibrar la demanda de energía y la producción en la célula [R].
También es responsable de iniciar la destrucción de células viejas y defectuosas, un proceso llamado apoptosis. Esto es necesario para hacer espacio para el crecimiento y la regeneración de las células después de las lesiones y para prevenir el crecimiento del cáncer [R, R, R].
Los subproductos de las actividades mitocondriales se acumulan y producen radicales libres. Esto causa estrés oxidativo, una causa principal de casi todas las enfermedades relacionadas con la edad [R].
La función mitocondrial inadecuada conduce a la acumulación de desechos y evita que las células defectuosas se eliminen del cuerpo. Esto puede resultar en el crecimiento del tumor y el cáncer [R].
Las mitocondrias son el objetivo de una serie de nuevas terapias prometedoras para el cáncer, la insuficiencia cardíaca, la resistencia a la insulina y los trastornos neurodegenerativos [R].
Dado que la energía es fundamental para la vida, mejorar la función mitocondrial está directamente relacionado con la salud física y mental.
ADN mitocondrial
La mayoría del genoma humano, que es todo el ADN de un individuo, está empaquetado en 23 pares de cromosomas en el núcleo de la célula. Además del ADN nuclear, los humanos también tienen un conjunto específico de 37 genes que solo se encuentran en la mitocondria [R].
El ADN mitocondrial (ADNmt) es un cromosoma único y circular. Cada mitocondria lleva de 2 a 10 copias [R].
El ADN mitocondrial codifica principalmente las proteínas que son necesarias para la función mitocondrial. En contraste, el ADN nuclear codifica las proteínas que se requieren para activar la síntesis de proteínas mitocondriales [R].
Los 23 pares de cromosomas en el núcleo se heredan de ambos padres después de someterse a una recombinación, un proceso que te hace único [R].
Por el contrario, el ADN mitocondrial solo puede heredarse por vía materna. Esto significa que el ADN mitocondrial se transmite únicamente de la madre. El ADNmt paterno se descompone y se desecha después de que el esperma ingresa al óvulo [R].
Origen de la mitocondria
Algunos de los primeros organismos vivos en la tierra fueron organismos procarióticos unicelulares como bacterias. Los seres humanos son organismos eucarióticos, lo que significa que estamos compuestos de células eucarióticas. Se plantea la hipótesis de que las células eucariotas se originaron a partir de células procariotas [R, R].
Los procariotas se diferencian de los eucariotas en varias formas [R]:
- Los procariotas no tienen compartimentos internos (orgánulos).
- El ADN es circular, no lineal como los eucariotas.
- Los procariotas no tienen un núcleo y, por lo tanto, su ADN flota libremente dentro del centro de la célula, mientras que el ADN eucariótico se empaqueta en el núcleo de cada célula.
- El ADN procariótico es más simple que el ADN eucariótico.
- Los procariotas se replican al dividirse por la mitad y producir 2 copias idénticas (fisión binaria).
Debido a sus muchas similitudes, se cree que la mitocondria ha evolucionado a partir de antiguas células procarióticas que usan oxígeno como las bacterias [R, R].
- Estructuralmente, la mitocondria se parece a las bacterias.
- Está compuesto por una doble membrana a través de la cual se sintetiza ATP, similar a la producción bacteriana de ATP.
- Lleva su propio ADN circular.
- La secuencia del ADN mitocondrial se parece mucho a la secuencia del ADN en los procariotas modernos (Rickettsia prowazekii).
- Al igual que los organismos procarióticos, la mitocondria se puede replicar dentro de la célula mediante fisión binaria.
La teoría más aceptada que explica estas características es la teoría endosimbiótica. Esta teoría sugiere que los antiguos organismos procarióticos que producían oxígeno mediante el uso de energía se fusionaron accidentalmente con células eucarióticas que no utilizaban oxígeno. Las dos células se benefician mutuamente de trabajar juntas como una unidad individual (endosimbiontes) [R, R].
La capacidad de usar oxígeno para obtener energía demostró ser favorable tanto para la supervivencia de las células. Con el tiempo, las células fusionadas evolucionaron en organismos eucarióticos modernos. Se cree que la mitocondria es un remanente del organismo procariótico original [R].
Estructura
La estructura de la mitocondria es altamente inusual, lo que es propicio para su función como máquina de fabricación de ATP [R].
La mitocondria está encerrada por una doble membrana y el interior del orgánulo se llama matriz. La membrana interna se pliega múltiples veces en estructuras llamadas crestas. Las proteínas productoras de ATP están ubicadas entre estas dos membranas y el movimiento de iones a través de la membrana interna potencia estas proteínas. La presencia de las crestas aumenta el área de superficie disponible para la producción de ATP [R, R].
La matriz contiene una copia del ADN circular y las proteínas responsables de mediar la actividad mitocondrial. Estas incluyen proteínas que crean ATP, estimulan la división mitocondrial y la fusión de las mitocondrias, metabolizan las moléculas e inducen la apoptosis [R].
Las mitocondrias pueden sufrir división (fisión) y fusión. La división mitocondrial aumenta el número de mitocondrias al dividir la mitocondria por la mitad o producir un pequeño fragmento de la mitocondria original [R].
La fusión mitocondrial ocurre cuando dos mitocondrios se fusionan, formando una estructura alargada. Esto suele ocurrir cuando hay daños. Al fusionarse, las mitocondrias pueden superar la disfunción permitiendo que las dos mitocondrias semi-dañadas funcionen como una mitocondria que funciona completamente (complementación) [R].
Mitocondria vs. Cloroplastos
La estructura y función de las mitocondrias son muy similares a los cloroplastos. Los cloroplastos se encuentran dentro de las plantas y algunas algas. Como las mitocondrias, ellas:
- tener su propia doble membrana
- Tienen su propio conjunto de ADN y ARN.
- proporcionar energía a la célula
- tienen conjuntos similares de enzimas y proteínas
A diferencia de las mitocondrias, los cloroplastos utilizan la luz solar para producir energía para la planta a través de un proceso conocido como fotosíntesis.
Función de las mitocondrias
La mitocondria es responsable de la conversión de grasas y carbohidratos en ATP, la moneda de energía de la célula. Además, las mitocondrias pueden inducir la muerte celular cuando sea necesario, gestionar los niveles de calcio y descomponer las moléculas basadas en carbono [R, R].
Los subproductos de la función mitocondrial normal se acumulan y producen radicales libres. Esto conduce al estrés oxidativo, que es la causa principal de casi todas las enfermedades graves relacionadas con la edad [R].
1) Las mitocondrias son la potencia de la célula
El papel más importante que desempeñan las mitocondrias es la producción de ATP. Una serie de reacciones bioquímicas tanto dentro como fuera de las mitocondrias dan como resultado la producción de ATP a través de un proceso llamado fosforilación oxidativa. Además de tener una función celular normal, estas reacciones requieren alimentos y oxígeno para la producción de energía [R].
Las grasas, los carbohidratos y las proteínas proporcionan al cuerpo diferentes cantidades de ATP cuando se descomponen [R].
Los carbohidratos, que se convierten en moléculas de glucosa, son los más rápidos de descomponer y generan de 34 a 38 moléculas de ATP por cada molécula de glucosa que se somete a la respiración. Son la forma de energía preferida del cuerpo [R].
Las grasas se utilizan para obtener energía cuando los niveles de glucosa son bajos. Una sola molécula de ácido palmítico, una grasa saturada común, puede producir 130 moléculas de ATP. Sin embargo, la descomposición de las grasas (lipólisis) es un proceso mucho más complejo, y puede demorar hasta 72 horas para digerir una comida grasa [R].
Las proteínas solo se usan para obtener energía cuando el cuerpo está muerto de hambre [R].
¿Qué es la respiración celular?
Hay 3 etapas en la respiración celular [R]: glucólisis, ciclo de Krebs y fosforilación oxidativa.
Glucólisis
Una vez que los carbohidratos o el glicerol de las grasas se descomponen en moléculas de glucosa, experimentan una serie de reacciones llamadas glucólisis [R].
- La glucólisis ocurre fuera de las mitocondrias, en el citosol de la célula.
- Es un proceso anaeróbico, lo que significa que no necesita oxígeno para ocurrir.
- Da como resultado la producción neta de 2 moléculas de ATP, 2 moléculas de NADH + y 2 moléculas de piruvato.
La glucólisis solo no genera suficiente energía para la célula, pero los productos de viajes glucólisis en la mitocondria donde pasan a través del ciclo de Krebs, también conocido como el ciclo tricarboxílico-ácido (TCA) o el ácido cítrico ciclo [R].
- El piruvato y el NADH se difunden en las mitocondrias (matriz).
- El piruvato sufre una reacción que genera 2 moléculas intermedias llamadas acetil-CoA.
- El dióxido de carbono se produce como un producto de desecho y se puede excretar a través de los pulmones.
Las grasas se convierten en glicerol y colas de ácidos grasos. El glicerol se puede descomponer en glucosa en el citosol, que luego ingresa a la vía de la respiración celular en la glucólisis. Las colas de ácidos grasos sufren una serie de reacciones a través de un proceso llamado beta- oxidación. Esto produce acetil-CoA, que puede ingresar a la respiración en la segunda etapa [R].
En caso de inanición (ausencia de carbohidratos y grasas), los aminoácidos de las proteínas se convierten en pequeñas moléculas basadas en carbono en las mitocondrias. Estas moléculas pueden entrar en la vía respiratoria en el ciclo de Krebs para producir ATP. Esto es dañino porque las proteínas utilizadas para proporcionar energía se toman de nuestros huesos, músculos y piel [R].
Ciclo de Krebs
El ciclo de Krebs es la etapa en la que la glucosa se convierte completamente en dióxido de carbono usando oxígeno (oxidación). Implica una serie de reacciones utilizando el acetil-CoA generado previamente. El acetil-CoA producido a partir de la descomposición de las grasas entra en la respiración en esta etapa. El objetivo del ciclo de Krebs es producir moléculas de alta energía que luego se pueden usar para generar una mayor cantidad de ATP [R].
- La acetil-CoA ingresa al ciclo de Krebs en la matriz mitocondrial.
- Cuatro reacciones que requieren oxígeno dan como resultado el rendimiento neto de 2 ATP, 6 NADH y 2 moléculas de FADH2.
NADH y FADH2 son moléculas de alta energía que proporcionan la mayor fuente de energía para la producción de ATP. Se utilizan en la tercera etapa de la respiración, la fosforilación oxidativa [R].
Fosforilación Oxidativa
Esta etapa de la respiración es la más importante, ya que genera la mayor cantidad de ATP. Aquí es donde la estructura de la mitocondria juega un papel importante. Además, este proceso es la razón por la que necesitamos oxígeno para sobrevivir [R, R].
Entre las membranas externas e internas de la mitocondria existe un pequeño espacio llamado espacio intermembrana. Un grupo de enzimas mitocondriales y complejos de proteínas se alinean en la membrana interna, formando una estructura llamada cadena de transporte de electrones. La fosforilación oxidativa ocurre en dos pasos: transporte de electrones y quimiosmosis [R]
Cadena de transporte de electrones [R]:
- Seis NADH y dos FADH2 experimentan reacciones químicas y donan sus electrones al primero de los complejos de proteínas en la cadena de transporte de electrones.
- NADH se convierte en NAD +.
- FADH2 se convierte en FAD.
- Los electrones pasan de complejo a complejo a lo largo de la cadena.
- A medida que los electrones pasan de un complejo de proteínas a otro, generan energía.
Quiosmosis [R]:
- La energía generada al pasar los electrones por la cadena de transporte se usa para bombear iones H + desde la matriz mitocondrial interna al espacio intermembrana.
- Los iones H + se acumulan entre las dos membranas mitocondriales.
- En algún punto, la cantidad de iones H + dentro del espacio de la membrana interna es mucho mayor que la cantidad de H + dentro de la matriz mitocondrial, creando un gradiente químico a través de la membrana interna.
- Los iones H + regresan a la matriz por su gradiente de concentración, liberando una gran cantidad de energía.
- La enzima final de la cadena de transporte de electrones, ATP sintasa, recoge la energía generada por el flujo de iones H + en la matriz.
- La ATP sintasa usa esta energía para generar de 34 a 38 moléculas de ATP y 6 moléculas de H2O.
El propósito del oxígeno en el proceso respiratorio es recoger los iones H + que fluyen de vuelta a la célula y forman moléculas de agua. Si no tuviéramos oxígeno, los iones H + permanecerían dentro de la matriz mitocondrial. Esto disminuiría el gradiente creado a través de la membrana interna porque habrá iones H + en ambos lados de la membrana. Esto evitará la quimiosmosis y. en última instancia, no habrá energía para la producción de ATP [R, R].
2) Las mitocondrias pueden desencadenar la muerte celular
Las mitocondrias desempeñan un papel importante en el inicio de la muerte celular cuando la célula está dañada o envejecida. Esto es útil para enfocar la producción de energía en células y tejidos que tienen mayores demandas de energía. La muerte celular también evita la propagación de células mutadas y defectuosas. El proceso de suicidio celular se llama apoptosis, que está mediado en gran medida por la liberación de proteínas “suicidas” de las mitocondrias [R, R].
Cuando solo se daña una mitocondria, las mitocondrias pueden destruirse a sí mismas, mediante un proceso llamado mitofagia, dejando la célula y otras mitocondrias intactas [R].
La cadena de transporte de electrones, que se encuentra en la membrana interna de la mitocondria, a menudo filtra radicales libres o especies reactivas de oxígeno (ROS). Si bien la producción de radicales libres es normal, puede provocar un estrés oxidativo dañino en la célula. Los radicales libres reaccionan con muchas otras sustancias en la célula, lo que lleva al daño al ADN y la destrucción de muchas proteínas vitales [R, R].
La acumulación de estrés oxidativo está vinculada a más de 200 enfermedades humanas. La apoptosis, o muerte celular programada, previene la acumulación de radicales libres. Las mitocondrias pueden responder a señales apoptóticas dentro (vía intrínseca) y fuera de la célula (vía extrínseca) [R, R].
Aparte de los radicales libres, ciertas proteínas pueden unirse a los receptores en la superficie de la célula e iniciar la apoptosis. Estas se llaman señales de muerte y se liberan en respuesta a mutaciones del ADN, radiación o falta de nutrientes [R, R].
Cuando las señales de muerte se unen a sus receptores en la célula, causan una serie de reacciones químicas que conducen a cambios en la membrana mitocondrial. Esto conduce al movimiento de proteínas hacia la matriz mitocondrial, liberando una proteína llamada citocromo C desde la membrana interna [R, R].
El citocromo C es una de las proteínas en la cadena de transporte de electrones. Cuando se libera de la mitocondria, la célula debe sufrir apoptosis. Es el factor determinante de la muerte celular [R, R].
Los eventos destructivos de la apoptosis son llevados a cabo por las enzimas caspasas. Las caspasas existen en su forma inactiva en cada célula. Cuando se libera el citocromo C, una serie de reacciones químicas activan las enzimas caspasas. Las caspasas cortan o cortan las diferentes proteínas de la célula. A medida que avanza el proceso, las caspasas llevan a la destrucción completa de los contenidos celulares [R].
3) Las mitocondrias juegan un papel central en la descomposición de la mayoría de las moléculas orgánicas
Las mitocondrias contienen enzimas que descomponen muchas moléculas diferentes para ser utilizadas como energía en la vía de la respiración celular. Estas enzimas también producen moléculas intermedias que se componen de un solo átomo de carbono. Estas moléculas basadas en 1 carbono son altamente reactivas y pueden interferir con las actividades celulares naturales [R, R].
Dentro de las mitocondrias, las moléculas de 1 carbono se convierten en diferentes aminoácidos que son necesarios para la producción de ATP y proteínas. Las vitaminas B9, B12, B6 y B2 proporcionan los carbonos necesarios para llevar a cabo estas reacciones. Las deficiencias en las vitaminas B están relacionadas con enfermedades relacionadas con la edad como la Enfermedad de Alzheimer, las enfermedades cardiovasculares y el cáncer [R, R].

La producción de grupos hemo, el núcleo estructural de la hemoglobina, que se encuentra en los glóbulos rojos, es un ejemplo de una sustancia importante producida por el metabolismo del carbono 1 en las mitocondrias. La hemoglobina es una proteína que contiene hierro que permite que el oxígeno y el dióxido de carbono se transporten a la sangre y se transporten por el cuerpo [R].
Las moléculas basadas en 1 carbono se exportan desde las mitocondrias para producir algunos de los componentes básicos del ADN. Esto ocurre en el fluido gelatinoso que llena el interior de una célula (citoplasma), donde los productos pueden moverse a diferentes partes de la célula según sea necesario o exportarse a otros tejidos [R].
Las mitocondrias también forman parte del ciclo de la urea, el proceso por el cual losátomos de nitrógeno de diferentes moléculas se excretan a través del cuerpo como urea. La urea es la sustancia principal que se encuentra en la orina y produce su color amarillo [R, R].
Por lo tanto, las mitocondrias evitan la interrupción de la función celular normal, participan en la eliminación de productos de desecho y proporcionan moléculas precursoras necesarias para otras reacciones.
4) Niveles de calcio de las mitocondrias
Las mitocondrias monitorean los niveles de calcio en la célula para coordinar la producción de energía con la demanda de energía. Los niveles de calcio aumentan en la célula para la activación de casi cualquier vía bioquímica. La activación de una célula, a su vez, requiere energía, por lo que las mitocondrias tendrán que aumentar la producción de ATP [R].
Cuando el calcio ingresa a la célula, la concentración de calcio aumenta fuera de la matriz mitocondrial. Esto crea un gradiente de concentración que impulsa el flujo de calcio a la matriz. Los canales de proteínas que permiten la entrada de calcio se encuentran en la membrana interna y se abren en respuesta a las altas concentraciones de calcio intracelular [R].
En las células musculares, la entrada de calcio produce un aumento de la producción mitocondrial. Esto permite que el suministro de energía satisfaga la demanda de energía de ese tejido [R].
Enfermedades vinculadas a la disfunción mitocondrial
Las mitocondrias que funcionan correctamente son fundamentales para la salud, ya que es el principal proveedor de energía de la célula. Sin embargo, las especies oxidativas reactivas producidas por las mitocondrias se acumulan con el tiempo y el estrés oxidativo conduce a enfermedades relacionadas con la edad. Debido al vasto papel de las mitocondrias en la célula, la disfunción mitocondrial está vinculada a cientos de enfermedades [R, R, R].
Además, una serie de trastornos metabólicos son causados por mutaciones genéticas en el ADN mitocondrial o nuclear. Estas mutaciones pueden ser heredadas u ocurrir aleatoriamente [R].
1) Cáncer y disfunción mitocondrial.
Las células cancerosas requieren mitocondrias para potenciar el crecimiento de tumores. Las células cancerosas tienen un mayor número de mitocondrias para proporcionar esta energía. El aumento mitocondrial está mediado independientemente por diferentes factores de transcripción o proteínas que inician la producción de genes específicos [R].
Por otro lado, las células cancerosas aumentan el volumen de negocios de las mitocondrias que acumulan radicales libres. El estrés oxidativo aumenta en las células cancerosas, lo que daña el tejido circundante [R].
Una de las características del cáncer es la capacidad de la célula para evitar la muerte celular programada (apoptosis). Normalmente, la mitocondria de las células sanas desencadenaría este proceso si la célula se replicara demasiado o demasiado rápido. Sin embargo, en las células cancerosas, la muerte celular programada se evita aumentando la destrucción de las mitocondrias que acumulan radicales libres. También activan las vías antioxidantes para que el estrés oxidativo no desencadene la muerte celular [R, R].
Las mitocondrias de las células cancerosas tienen niveles más bajos de proteínas que promueven la muerte celular (BAX / BAK) y / o niveles más altos de proteínas que previenen la muerte celular (BCL-2 / BCL-XL) [R, R].
La red de mitocondrias en las células cancerosas también es diferente. Las células cancerosas tienen mitocondrias más fragmentadas (al aumentar la división mitocondrial y disminuir la fusión) [R].
Las células cancerosas también producen energía de manera diferente a través de un proceso conocido como el efecto Warburg. La producción de energía se realiza en gran parte sin oxígeno (anaeróbicamente), a través de la glucólisis. Esto puede deberse a la reducción de la función mitocondrial para evitar la apoptosis [R, R].
La respiración aeróbica a través del ciclo de Krebs y la fosforilación oxidativa todavía ocurren en las células cancerosas, pero en menor grado.
2) Enfermedades neurodegenerativas y disfunción mitocondrial
La disfunción mitocondrial es una causa importante de enfermedades neurodegenerativas relacionadas con la edad como el Alzheimer, la ELA y la Enfermedad de Parkinson. La acumulación de radicales libres con la edad da como resultado el daño al ADN y a las proteínas. Las mitocondrias acumulan proteínas defectuosas que causan la pérdida de producción de energía y, en última instancia, la muerte celular [R].
Enfermedad de Alzheimer
- Las proteínas beta amiloides se acumulan alrededor de la membrana mitocondrial externa.
- Esta acumulación disminuye la producción de ATP, aumenta el estrés oxidativo y, en última instancia, causa la muerte celular.
- La beta amiloide aumenta la producción de proteínas mitocondriales.
- Las enzimas mitocondriales tienen actividad disminuida, lo que lleva a niveles reducidos de ATP.
- Las mitocondrias sufren cambios estructurales dentro de la célula. En lugar de existir en tubos largos (fusión mitocondrial), las mitocondrias se fragmentan en pequeñas piezas dentro de la célula (fisión). Esto se suma a la disfunción general de las células cerebrales observadas en pacientes con Enfermedad de Alzheimer [R].
Enfermedad de Parkinson
- El sello distintivo de la Enfermedad de Parkinson es la acumulación de proteína alfa-sinucleína, que conduce a la muerte celular y la pérdida de neuronas.
- Los pacientes con Parkinson acumulan esta proteína en la mitocondria, lo que lleva a un aumento del estrés oxidativo y reduce la producción de energía.
- La proteína parkin (E3 ubiquitin ligase) y la proteína PINK1 son responsables de marcar las mitocondrias dañadas para su destrucción. Los pacientes con Enfermedad deParkinson tienen niveles bajos de estas proteínas.
- Las mitocondrias dañadas y de bajo funcionamiento no se degradan y permanecen en la célula.
- La alfa-sinucleína continúa acumulándose, lo que lleva a la neurodegeneración [R].
3) Diabetes y disfunción mitocondrial
La función mitocondrial inadecuada se observa en pacientes con diabetes tipo 1 y tipo 2.
Además de tener una falta de glucosa para la respiración, la red y la forma de las mitocondrias en las células de los pacientes diabéticos son anormales [R, R].
Las mitocondrias se dividen en pequeñas redes fragmentadas (mayor división, disminución de la fusión) en toda la célula. Esto se ve tanto en la diabetes tipo 1 como en la diabetes tipo 2 [R].

La diabetes tipo 2 es causada por la resistencia a la insulina en las células, lo que reduce la cantidad de glucosa disponible para la respiración. Esto disminuye la producción de ATP y, por lo tanto, la energía disponible para la celda [R].
Se desconoce si la disfunción mitocondrial es una causa de resistencia a la insulina o un síntoma de ella. Algunas pruebas apuntan a que las mitocondrias son una causa de resistencia a la insulina, en lugar de un síntoma [R].
Los pacientes con diabetes tipo 2 tienen niveles reducidos de proteínas mitocondriales responsables de la producción de energía [R, R].
4) Insuficiencia cardíaca y disfunción mitocondrial
Las células del corazón dependen en gran medida de las mitocondrias para impulsar el bombeo del corazón. La disfunción mitocondrial está implicada en la insuficiencia cardíaca debido a la acumulación de estrés oxidativo [R].
Los pacientes con insuficiencia cardíaca exhiben una actividad mitocondrial reducida. Las mitocondrias tienen menor actividad en las cadenas de transporte de electrones. Esto se debe a una pérdida de suministro de oxígeno a las mitocondrias [R].
Dado que el suministro de oxígeno se reduce, los electrones en la cadena de transporte de electrones no pueden ser recogidos por el oxígeno. Esto conduce a la acumulación de electrones, que producen radicales libres [R].
5) Síndrome de Fatiga Crónica y disfunción mitocondrial
El Síndrome de Fatiga Crónica, comúnmente conocido como SFC, es un trastorno de por vida caracterizado por síntomas prolongados (> 6 meses) de fatiga intensa que pueden reducir el funcionamiento diario en más del 50%. Los pacientes con SFC sufren de una variedad de otros síntomas, incluyendo [R]:
- Ansiedad
- Depresión
- Dolores de cabeza
- Dolor muscular
- Disfunción congnitiva
Aunque alguna vez se pensó que era una enfermedad mental, el aumento de la evidencia apunta a la disfunción mitocondrial como una de las principales causas de este trastorno [R].
Se han realizado múltiples ensayos clínicos y han producido resultados mixtos. El SFC puede ser causado por una o más de las siguientes anomalías mitocondriales [R])
- Forma mitocondrial más pequeña y número
- Niveles bajos de L-carnitina, ALCAR, ubiquinona o CoQ10
- Actividad proteica reducida en la cadena de transporte de electrones (fosforilación oxidativa)
- Reducción de la producción de ATP.
Varios estudios indicaron que no hubo diferencias significativas en la estructura o función mitocondrial de pacientes sanos y normales. Se requieren estudios adicionales para comprender completamente la causa de esta afección, el papel de la disfunción mitocondrial y cómo tratar a los pacientes de manera efectiva.
6) Trastornos genocondriales genéticos
Las mutaciones genéticas en los genes mitocondriales pueden resultar en disfunción mitocondrial a través de 1 o más de 5 mecanismos distintos [R]:
- Incapacidad para utilizar otras moléculas (sustratos) para la producción de energía
- Ciclo de Krebs inadecuado
- Producción de energía defectuosa a través de la cadena de transporte de electrones (fosforilación oxidativa)
- Transporte defectuoso de moléculas dentro y fuera de las mitocondrias
- Proteínas defectuosas en la cadena de transporte de electrones.
Estos defectos pueden ser causados por mutaciones en el ADN mitocondrial y / o nuclear. Además, pueden producirse defectos cuando el ADN mitocondrial no puede comunicarse con el ADN nuclear [R].
Algunas enfermedades metabólicas causadas por mutaciones del ADN mitocondrial y / o nuclear incluyen [R, R]:
- Síndrome de agotamiento del ADNmt (MDS)) MDS se refiere a un grupo de trastornos, cualquiera de los cuales tiene ADN mitocondrial disfuncional. Esto puede dar lugar a diferentes anomalías en el desarrollo, los músculos y el cerebro. Muchas enfermedades mitocondriales conocidas son un resultado de MDS.
- Miopatía mitocondrial) una enfermedad mitocondrial que causa problemas musculares como debilidad, intolerancia al ejercicio, dificultad para respirar o problemas con la visión.
- Encefalomiopatía mitocondrial, acidosis láctica y episodios similares a accidentes cerebrovasculares (MELAS, por sus siglas en inglés)) un trastorno mitocondrial que afecta el cerebro y los músculos en todo el cuerpo. Los episodios similares a un accidente cerebrovascular y la acumulación de ácido láctico dan como resultado demencia, vómitos, dolor extremo y debilidad muscular.
- Deficiencia de CoQ10) una deficiencia en la coenzima Q10, una proteína que forma parte de la cadena de transporte de electrones.
- Encefalomopatía neurogastrointestinal mitocondrial (MNGIE, por sus siglas en inglés)) una rara enfermedad mitocondrial que afecta principalmente el cerebro y el sistema digestivo. Los músculos y los nervios del sistema digestivo no empujan adecuadamente los alimentos a través del sistema.
- Diabetes mitocondrial: denominada diabetes y sordera hereditaria (MIDD), un subtipo de diabetes causada por una mutación única en el ADN mitocondrial (en la posición 3243). La enfermedad resulta en pérdida de audición y diabetes similar a la del tipo 1.
- Mutaciones POLG) un gen que codifica la subunidad gamma de la polimerasa del ADN, la parte activa (catalítica) de la proteína mitocondrial responsable de la síntesis del ADN. Las mutaciones en este gen conducen a la producción defectuosa de proteínas mitocondriales y muchas enfermedades mitocondriales.
El efecto del cuello de botella mitocondrial
Llevar mutaciones en su ADN mitocondrial no significa necesariamente que transmitirá la enfermedad. La proporción de células que portan mitocondrias mutadas tiene que ser mayor que las células portadoras de mitocondrias sanas para que la enfermedad se vea [R].
Durante la producción de células óvulos femeninas (ovocitos), cada célula ovárica llevará una selección aleatoria de copias de ADN mitocondrial. Algunas de las copias pueden tener mutaciones, mientras que otras pueden ser completamente normales. A medida que la célula del huevo madura y se prepara para la fertilización, muchas mitocondrias se replican al azar. Esto distribuye la posibilidad de heredar mitocondrias mutantes [R].
Este fenómeno significa que si la madre tiene mitocondrias altamente mutantes, su descendencia no necesariamente tendrá el rasgo [R].
Si suficientes células transportan ADN mitocondrial mutante, por ejemplo, más del 50% de las células en el cuerpo, es probable que el niño tenga el trastorno asociado.
Previniendo la herencia de ADN mitocondrial disfuncional (ADNmt)
Las nuevas tecnologías pueden prevenir la transmisión de ADN mitocondrial mutado de la madre a la descendencia. Una nueva técnica, llamada fertilización in vitro de 3 padres, consiste en cambiar el ADN nuclear de la madre con el ADN nuclear de un óvulo hembra donante que tiene un ADN mitocondrial sano. Por lo tanto, el óvulo del donante transporta la información genética de la madre, pero carece del ADN mitocondrial mutado que también porta.
Al utilizar la fertilización in vitro, el óvulo se insemina artificialmente con el semenpaterno. Luego, el óvulo fertilizado se reintroduce en el útero de la madre, donde puede adherirse al útero [R].
La descendencia tendrá todas las características físicas de sus padres biológicos porque el ADN nuclear no se modifica. Lo único diferente es que este niño tendrá una función mitocondrial adecuada, a diferencia de la madre que porta copias mutadas del ADN mitocondrial [R].
Puede tener disfunción mitocondrial si
- Te sientes excesivamente cansado [R]
- No puede hacer ejercicio durante largos períodos de tiempo [R]
- Se siente sin aliento, especialmente durante el ejercicio [R, R]
- Tienes mal crecimiento óseo y salud [R]
- Tiene dificultad para controlar sus movimientos, equilibrio y coordinación (ataxia) [R]
- Tienes problemas para caminar o hablar [R]
- Tienes debilidad muscular y dolor [R]
- Tiene una Enfermedad del músculo cardíaco (miocardiopatía) [R]
- Tienes problemas intestinales y digestivos [R]
- Tiene una Enfermedad del hígado y del riñón [R]
- Tiene párpados caídos, pérdida de visión y otros problemas oculares [R]
- Tiene diabetes y otros trastornos hormonales [R, R]
- Tiene problemas para escuchar [R]
- Experimenta más migrañas, accidentes cerebrovasculares y convulsiones [R, R]
- Tienes dificultad para recordar cosas [R]
- Tienes un retraso en el desarrollo [R]
- Eres autista [R]
- Tiene infecciones recurrentes [R, R]

El Boro como suplemento: beneficios y niveles

Interleucina 8 (IL-8): su relación con el cáncer y enfermedad del corazón
Transposición de las grandes arterias (TGA) – una anomalía congénita del corazón
Sobre el autor
saludybelleza
Rafa Montes, editor de Salud y Belleza. Investigación de temas de salud, bienestar, psicología y más. Puedes encontrarme en LinkedIn